Learning objectives
Participants first will learn about limitations of linear reference-based methods and work through a brief refresher or introduction to standard approaches for processing sequencing data, including read alignment and variant calling. Provided these motivating examples, we will use data from a variety of relevant sources to develop an intuition about pangenomic methods and a practical familiarity with applicable tools.
We will mostly use vg and other methods that read and write GFA.
For our exploration we have collected several data sets that combine genomic and phenotypic information, with a focus on contexts that are difficult to approach using standard linear reference methods:
- Bacteria (with resistance phenotypes) http://www.nature.com/articles/nmicrobiol201641
- HLA typing, e.g. from ONT data (tobi can ask whether we can use data from Wigard Klosterman)
- Yeast (with growth phenotypes) NCYC collection TODO Erik: get data from Ignacio
- Viral pooled population studies (HPV?)
Using these data we work through various modules based on different functional aspects of pangenomics.
This figure, from a recent paper on computational pangenomics, can serve as a helpful guide to our learning objectives:
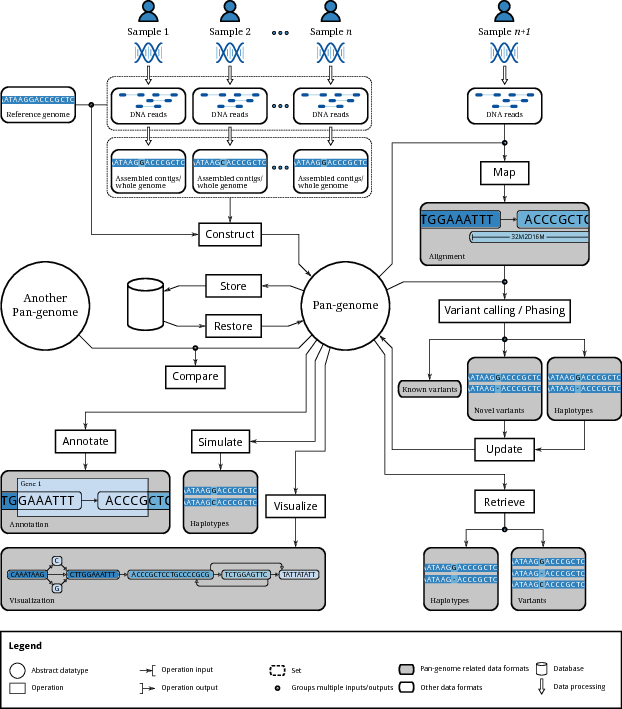
Our goal is that students achieve fluency in the practical aspects of relevant workflows within this scope.
Appreciate the limitations of linear reference genomes
Objectives:
- Know what a FASTA reference is, what a VCF or variant list is.
- Understand when we cannot simply align reads to a single reference and hope to correctly infer the unknown genome from which these reads are derived.
- Appreciate standard approaches to mitigate this issue.
Exercises:
- Align reads to a linear reference using bwa mem and call variants using freebayes.
- Align reads from a structural variant containing locus to a linear reference, visualize with IGV, and report what is found.
- Attempt to call variants in the region of a structural variant treated this way and discuss the results.
Construct
Objectives:
- Be able to describe a simple graphical model used for pangenome representation (vg’s data model).
- Understand the relationship between traditional lists of variants, linear references, and basic pangenomic models.
- Have a basic understanding of the graphical models used by assemblers and how they relate to the data model.
Linear reference + variants
Exercises:
- Generate a short paragraph summarizing the vg schema (using the schema itself, help from the instructors, and online documentation).
- Build a vg graph for a fragment of the human 1000 Genomes Project data using
vg construct.
Assembly graphs
Exercises:
- Run an assembler on a trivial (but real) data set and visualize the result with Bandage. (TODO: which data?)
- Write a script to count the contigs and edges in a GFA format output from this assembly process.
Collections of haplotypes
Exercises:
- Apply
vg msgaandcactusto assemble a graph from a fragment of the human MHC.
Visualize (bandage, tube maps, dot)
Objectives:
- Know basic techniques to visualize the sometimes-unweily pangenome systems.
Exercises:
Take graphs generated in our previous exercises and visualize them with:
- Bandage
- vg view
- IVG (we’ll return to this later when we do alignments)
Read mapping
Objectives:
- Understand the basic principles behind read alignment, and how these are complicated by aligning reads to a graph.
- Have the ability to make alignments and interrogate the result to learn basic information like read coverage or putative variants.
Exercises:
- TODO
Variant calling
TODO
(bonus) association study or population study
Back
Back to main page