Align HTS data against a genome
10.1 - Use the BWA aligner to align HTS data against a genome
One of the most common applications of NGS is resequencing, where we want to genotype an individual from a species whose genome has already been assembled (a reference genome), such as the human genome, often with the goal to identify mutations that can explain a phenotype of interest.
After obtaining millions of short reads, we need to align them to a (sometimes large) reference genome. To achieve this, novel, more efficient, alignment methods had to be developed. One popular method is based on the burrows-wheeler transform and the use of efficient data structures, of which bwa and bowtie are examples. They enable alignment of millions of reads in a few minutes, even in a laptop.
NOTE: Aligners based on the burrows-wheeler transform makes some assumptions to speed up the alignment process. Namely, they require the reference genome to be very similar to your sequenced DNA (less than 2-5% differences). Moreover, they are not optimal, and therefore sometimes make some mistakes.
TASK The first step of a burrows-wheeler aligner is to make an index from the fasta of the reference genome. open a terminal window, go to the folder resequencing (using the ‘cd’ command) and type bwa index NC_000913.3_MG1655.fasta. Now, we can do the alignment against the created database, type bwa mem NC_000913.3_MG1655.fasta SRR1030347_1.fastq.interval.fq SRR1030347_2.fastq.interval.fq > SRR1030347.alignment.sam.
NOTE: You may have noticed that we used paired fastq files in this alignment. The aligners can use the pairing information to improve the alignments, as we will see later.
10.2 - The SAM/BAM alignment format
To store millions of alignments, researchers also had to develop new, more practical formats. The Sequence Alignment/Map (SAM) format is a tabular text file format, where each line contains information for one alignment.
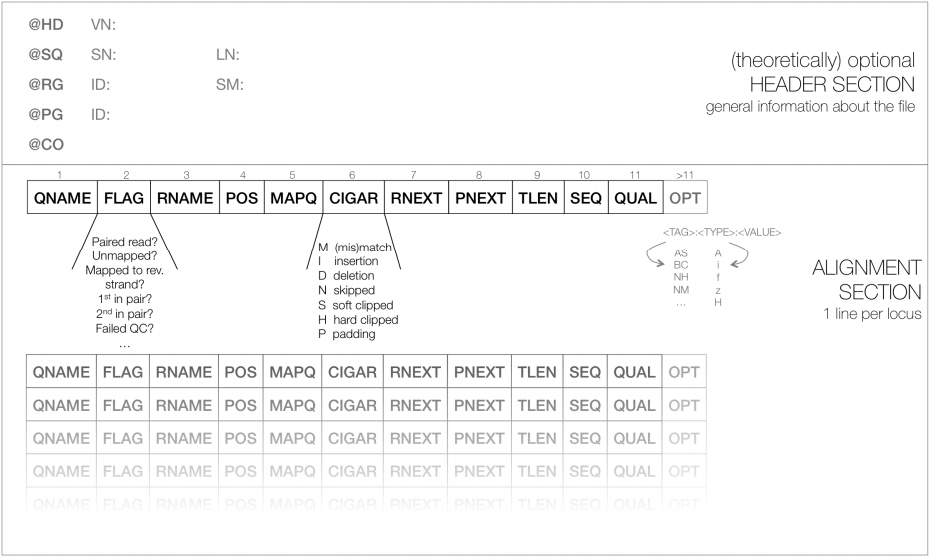
TASK Open the sam file you generated before (SRR1030347.alignment.sam) with a text editor, and/or type in the terminal window head SRR1030347.alignment.sam, making it easier to analyse/import the sam file into a spreadsheet program.
QUESTION: What is the position of the start of the first alignment in your SAM file?
Click Here to see the answer
Read SRR1030347.285 aligns starting in position 14 (information in the 4th column of the SAM).SAM files are most often compressed as BAM (Binary SAM) files, to reduce space. These BAM files can then be indexed (do not confuse this indexing with the indexing of the reference genome) to allow direct access to alignments in any arbitrary region of the genome. Several tools only work with BAM files.
TASK Let’s transform the SAM file into an indexed BAM file. In the same terminal window where you indexed the genome, type samtools view -Sb SRR1030347.alignment.sam > SRR1030347.alignment.bam. To create the index. The alignments in the bam file need to be sorted by position, type samtools sort SRR1030347.alignment.bam SRR1030347.alignment.sorted. Finally, we can create the index, by typing samtools index SRR1030347.alignment.sorted.bam.
Notice now the appearance of a companion file SRR1030347.alignment.sorted.bam.bai that contains the index. This file should always accompany its corresponding bam file.
TASK Let’s do the whole process using galaxy. Upload the reference genome NC_000913.3_MG1655.fasta and the paired fastq files SRR1030347_1.fastq.interval.fq and SRR1030347_2.fastq.interval.fq into Galaxy. Check the fastq quality and perform any necessary filtering using trimmomatic or with any of the tools we saw before. Next, perform an alignment with bwa mem of the paired reads (you need to select the option of paired reads) against the reference genome (choose one from history). Next, download the bam file that was created. Also, download the companion bai index file (you need to press on the download icon to have the option to download the bam and the bai files).
NOTE: Assess how well you achieve the learning outcome. For this, see how well you responded to the different questions during the activities and also make the following questions to yourself.
-
Did you broadly understand the challenges of aligning millions of short reads to a genome?
-
Did you broadly understand the assumptions underlying the use of burrows-wheeler aligners?
-
Could you use bwa to align reads to a reference genome?
-
Do you know what is the most common alignment format these aligners use?
-
Do you broadly understand the contents of a SAM/BAM file and the difference between SAM and BAM?
Back
Back to main page.