Proteomics assay data
Proteomics assay data
Assays for high-throughput proteomics have recently become available, allowing the screening of thousands of proteins in large number of samples. Two technologies are currently widely encountered: aptamers and antibodies (ref). Both provide data in a tabular format with the protein abundance estimates for every sample.
Libraries
We will need the following libraries. If they are not availble on your
system, use the ìnstall_packages command to install them.
library(janitor) # A package to clean dirty data
##
## Attaching package: 'janitor'
## The following objects are masked from 'package:stats':
##
## chisq.test, fisher.test
library(tidyr) # A package to tidy data
library(dplyr) # A package to manipulate data
##
## Attaching package: 'dplyr'
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
library(parallel) # A package to run tasks in parallel
library(glue) # A package to glue variables in strings
library(stringr) # A package to manipulate strings
library(ggplot2) # A package to plot data
library(scico) # A package of scientific color palettes
library(conflicted) # A package to manage namespace conflicts
# Resolve conflicting function names
conflict_prefer("filter", "dplyr")
## [conflicted] Will prefer dplyr::filter over any other package
# Set the theme for plots
theme_set(theme_bw(base_size = 13))
Load data
:pencil2: Open the data set available at these links: - link1 - link2 - link3
files_folder <- "/home/marc/Projects/tutorials/data"
protein_values <- read.table(
file = file.path(files_folder, "soma_example.gz"),
header = T,
sep = "\t",
stringsAsFactors = F
) %>%
clean_names() # This function makes the formatting of the column names uniform, note that it transforms the identifiers of the proteins
# write.table(
# x = protein_values,
# col.names = T,
# row.names = F,
# sep = "\t",
# file = file.path(files_folder, "soma_example.gz")
# )
samples_details <- read.table(
file = file.path(files_folder, "soma_example_2.gz"),
header = T,
sep = "\t",
stringsAsFactors = F
) %>%
clean_names()
protein_details <- read.table(
file = file.path(files_folder, "soma_example_3.gz"),
header = T,
sep = "\t",
stringsAsFactors = F,
comment.char = ""
) %>%
clean_names() %>%
mutate(
protein_id = paste0("x", str_replace_all(string = seq_id, pattern = "-", replacement = "_")) # Note that we create a column to match identifiers with the column names of the protein_values
)
:speech_balloon: What is shown in row/columns in each file? How many proteins were measured in how many samples?
Abundance estimates standardization
:pencil2: Select a sample and make a histogram of the protein abundance estimates.
selected_sample <- sample(samples_details$id, 1)
selected_sample_values <- protein_values %>%
filter(
sample_id == selected_sample
) %>%
select(
starts_with("x")
) %>%
pivot_longer(
cols = starts_with("x")
)
ggplot() +
geom_histogram(
data = selected_sample_values,
mapping = aes(
x = value
),
bins = 50,
col = "darkblue",
fill = "darkblue",
alpha = 0.4
) +
scale_x_continuous(
name = glue("Abundance estimate for sample {selected_sample}")
) +
scale_y_continuous(
name = glue("Number of proteins"),
expand = expansion(
mult = c(0, 0.05)
)
)
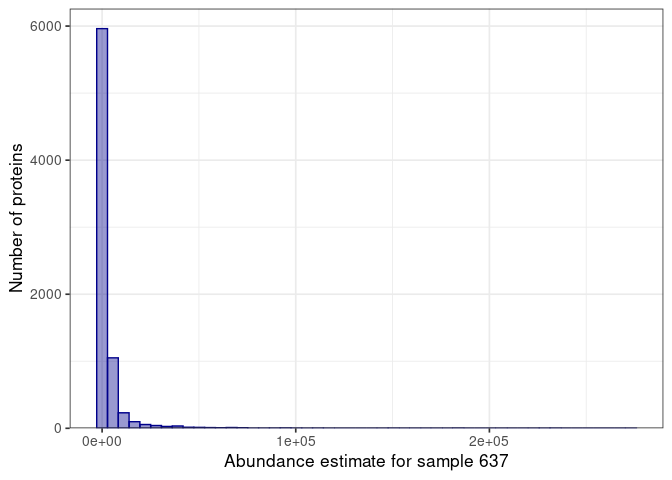
:speech_balloon: Is this what you would expect when looking at the proteome of a sample?
:pencil2: Select a protein and look at the distribution of abundance estimates.
selected_protein <- sample(protein_details$protein_id, 1)
ggplot() +
geom_histogram(
data = protein_values,
mapping = aes(
x = !!sym(selected_protein)
),
bins = 50,
col = "darkgreen",
fill = "darkgreen",
alpha = 0.4
) +
scale_x_continuous(
name = glue("Abundance estimate of {selected_protein}")
) +
scale_y_continuous(
name = glue("Number of samples"),
expand = expansion(
mult = c(0, 0.05)
)
)
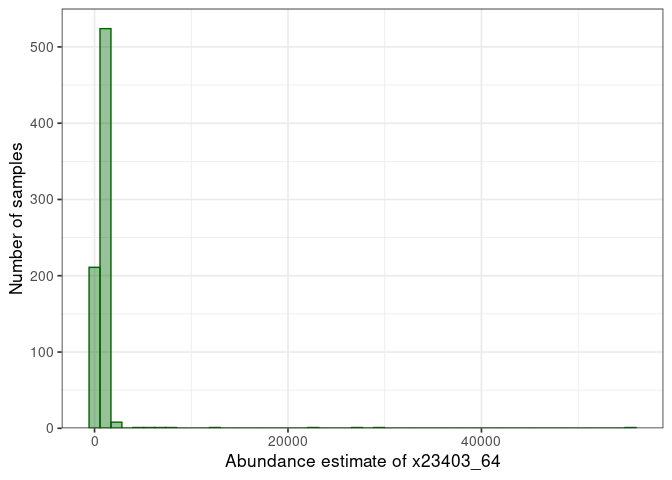
:speech_balloon: How does the distribution looks like? How will it influence downstream analyses?
:pencil2: Log, center, and scale the abundance of each protein.
for (protein in protein_details$protein_id) {
log_values <- log10(protein_values[[protein]][protein_values$sample_id %in% samples_details$id & !is.na(protein_values[[protein]]) & protein_values[[protein]] > 0])
if (length(log_values) < 10) {
stop(glue("Too few values for protein {protein}"))
}
mean_log <- mean(log_values, na.rm = T)
sd_log <- sd(log_values, na.rm = T)
new_column <- paste0(protein, "_log_std")
protein_values[[new_column]] <- ifelse(protein_values$sample_id %in% samples_details$id & !is.na(protein_values[[protein]]) & protein_values[[protein]] > 0, (log10(protein_values[[protein]]) - mean_log) / sd_log, NA)
}
:pencil2: Plot again the distribution for the selected protein.
selected_protein_log_std <- paste0(selected_protein, "_log_std")
ggplot() +
geom_histogram(
data = protein_values,
mapping = aes(
x = !!sym(selected_protein_log_std)
),
bins = 50,
col = "darkgreen",
fill = "darkgreen",
alpha = 0.4
) +
scale_x_continuous(
name = glue("Standardized abundance estimate of {selected_protein}")
) +
scale_y_continuous(
name = glue("Number of samples"),
expand = expansion(
mult = c(0, 0.05)
)
)
## Warning: Removed 3 rows containing non-finite values (stat_bin).
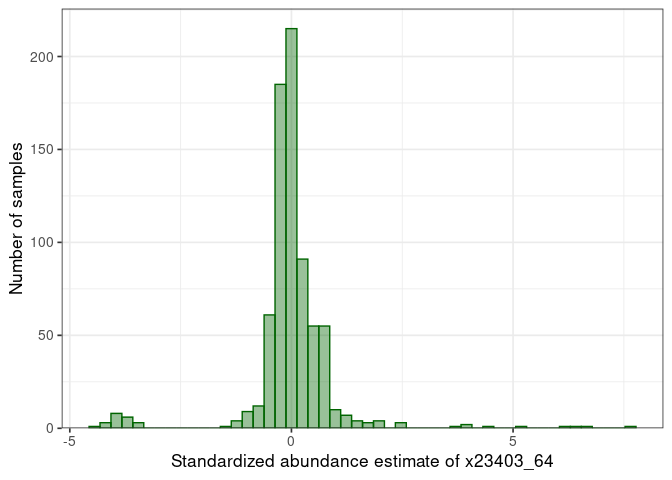
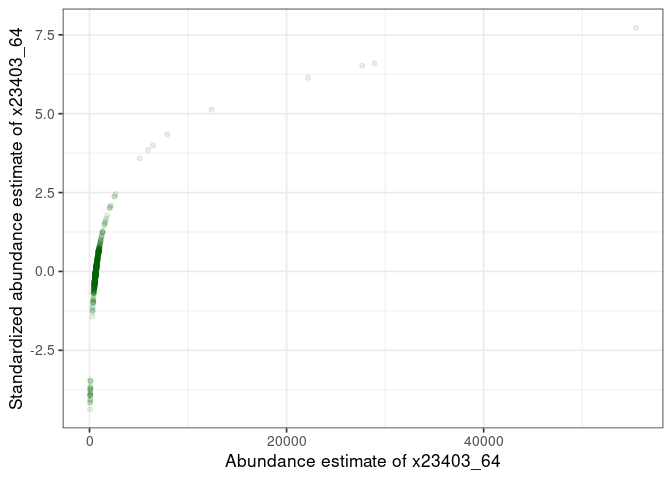
:pencil2: Plot the standardized vs non-standardized values.
ggplot() +
geom_point(
data = protein_values,
mapping = aes(
x = !!sym(selected_protein),
y = !!sym(selected_protein_log_std)
),
col = "darkgreen",
alpha = 0.1
) +
scale_x_continuous(
name = glue("Abundance estimate of {selected_protein}")
) +
scale_y_continuous(
name = glue("Standardized abundance estimate of {selected_protein}")
)
## Warning: Removed 3 rows containing missing values (geom_point).
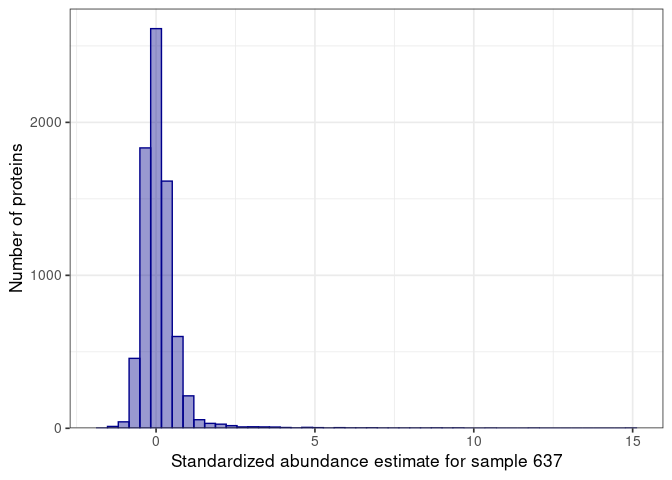
:pencil2: Plot again the distribution for the selected sample.
selected_sample_std_values <- protein_values %>%
filter(
sample_id == selected_sample
) %>%
select(
ends_with("_log_std")
) %>%
pivot_longer(
cols = ends_with("_log_std")
)
ggplot() +
geom_histogram(
data = selected_sample_std_values,
mapping = aes(
x = value
),
bins = 50,
col = "darkblue",
fill = "darkblue",
alpha = 0.4
) +
scale_x_continuous(
name = glue("Standardized abundance estimate for sample {selected_sample}")
) +
scale_y_continuous(
name = glue("Number of proteins"),
expand = expansion(
mult = c(0, 0.05)
)
)
:pencil2: Plot the standardized vs non-standardized values.
plot_values <- merge(selected_sample_values, selected_sample_std_values %>% rename(std_value = value) %>% mutate(name = str_remove(name, "_log_std")), by = "name")
ggplot() +
geom_point(
data = plot_values,
mapping = aes(
x = value,
y = std_value
),
col = "darkblue",
alpha = 0.1
) +
scale_x_continuous(
name = glue("Protein abundance estimate for {selected_sample}")
) +
scale_y_continuous(
name = glue("Standardized abundance estimate for {selected_sample}")
)
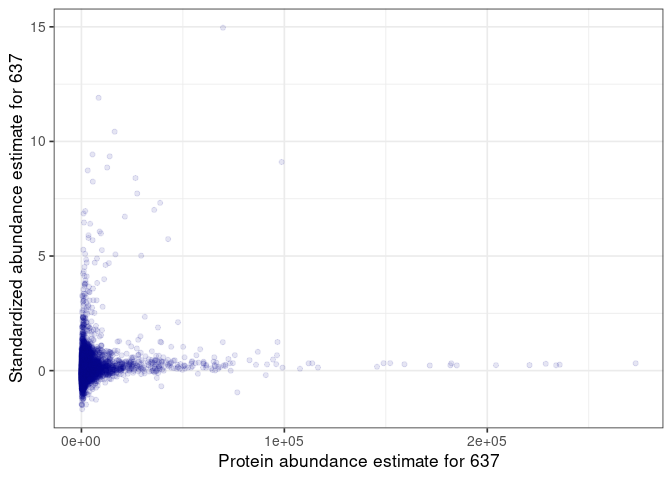
:speech_balloon: How do you interpret this plot?
Principal component analysis
:pencil2: Run a PCA on the standardized values.
pca_input <- protein_values %>%
filter(
sample_id %in% samples_details$id
) %>%
select(
sample_id, ends_with("_log_std")
)
pca_samples <- pca_input$sample_id
pca_input <- t(pca_input[, -1])
colnames(pca_input) <- pca_samples
pca <- prcomp(pca_input)
The quantitative information is now projected in a base of dimensions ordered by decreasing amount of variance, the principal components. We can plot the dimensions capturing most of the variance from the sdev attribute of the pca result.
:pencil2: Plot the variance explained by the first ten components
eigen_values <- pca$sdev ^ 2
total_eigen_value <- sum(eigen_values)
pc_contribution <- 100 * eigen_values / total_eigen_value
ggplot() +
geom_col(
mapping = aes(
x = 1:10,
y = pc_contribution[1:10]
)
) +
scale_x_continuous(
name = "PC"
) +
scale_y_continuous(
name = glue("Proportion of Variance [%]"),
expand = expansion(
mult = c(0, 0.05)
)
)
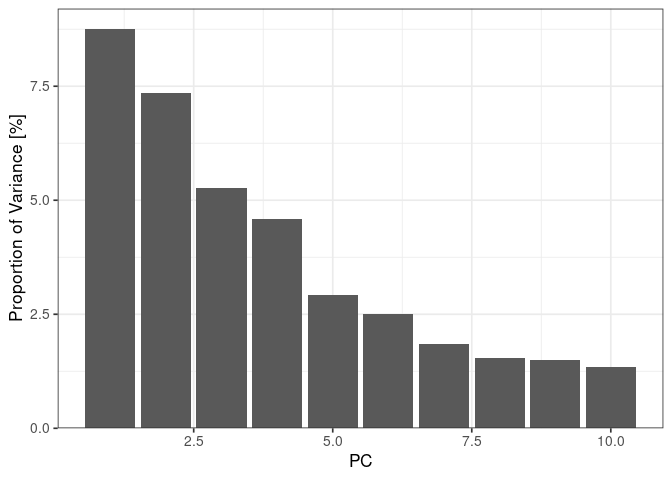
:speech_balloon: How do you interpret this plot?
:pencil2: Plot the samples in the plane made by PC1 and PC2
pc1 <- pca$rotation[,1]
pc2 <- pca$rotation[,2]
names <- dimnames(pca$rotation)[[1]]
plot_data <- data.frame(
pc1 = pc1,
pc2 = pc2,
id = names
)
contribution1 <- round(100 * eigen_values[1] / total_eigen_value)
contribution2 <- round(100 * eigen_values[2] / total_eigen_value)
ggplot() +
geom_point(
data = plot_data,
mapping = aes(
x = pc1,
y = pc2
),
alpha = 0.2
) +
xlab(glue("PC1 [{contribution1}%]")) +
ylab(glue("PC2 [{contribution2}%]"))
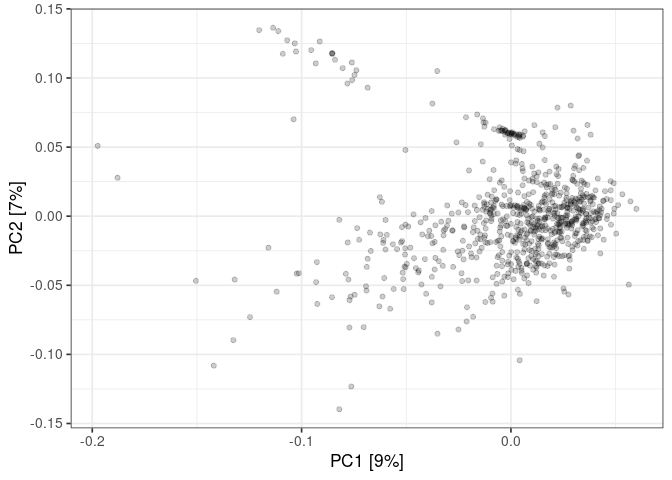
:pencil2: Color the points using the sample details.
plot_data <- plot_data %>%
left_join(
samples_details %>%
mutate(
id = as.character(id)
),
by = "id"
)
ggplot() +
geom_point(
data = plot_data,
mapping = aes(
x = pc1,
y = pc2,
col = age
),
alpha = 0.2
) +
xlab(glue("PC1 [{contribution1}%]")) +
ylab(glue("PC2 [{contribution2}%]")) +
scale_color_scico(
name = "Age",
palette = "berlin"
)
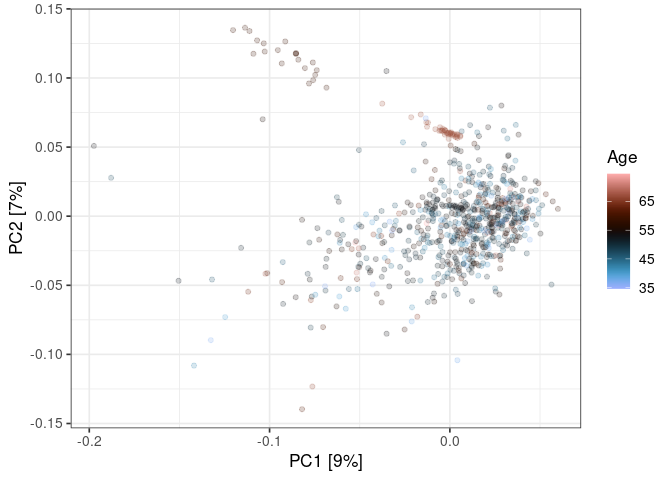
ggplot() +
geom_point(
data = plot_data,
mapping = aes(
x = pc1,
y = pc2,
col = bmi
),
alpha = 0.2
) +
xlab(glue("PC1 [{contribution1}%]")) +
ylab(glue("PC2 [{contribution2}%]")) +
scale_color_scico(
name = "BMI",
palette = "berlin"
)
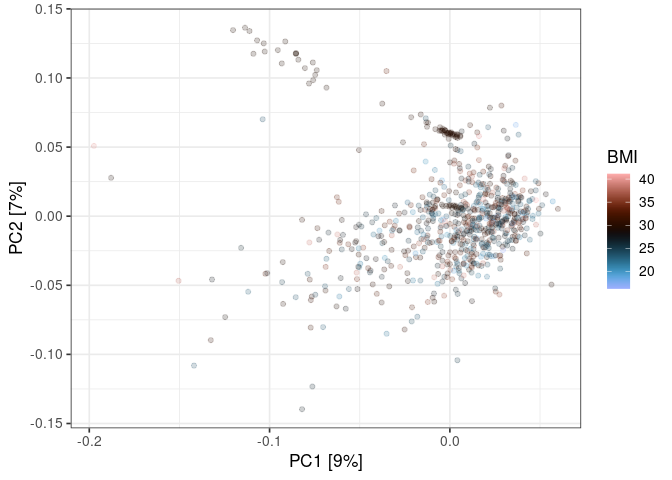
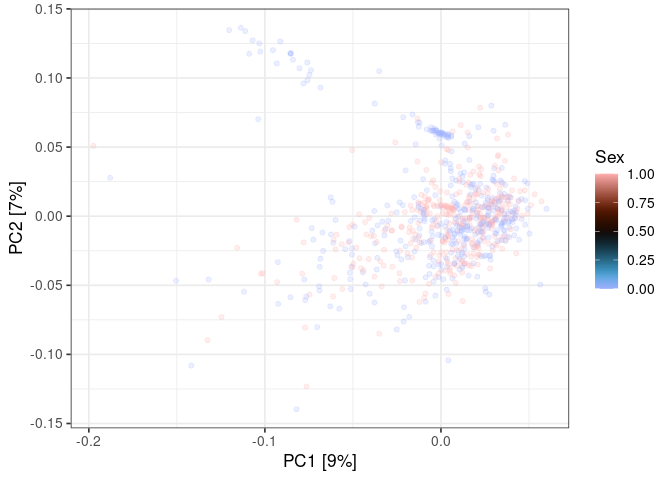
ggplot() +
geom_point(
data = plot_data,
mapping = aes(
x = pc1,
y = pc2,
col = sex
),
alpha = 0.2
) +
xlab(glue("PC1 [{contribution1}%]")) +
ylab(glue("PC2 [{contribution2}%]")) +
scale_color_scico(
name = "Sex",
palette = "berlin"
)
Association analysis
:pencil2: Compute the association of the selected protein with one of the disease classes
class <- "Class_1"
samples_tested <- samples_details %>%
filter(
group == "Healthy" | group == class
) %>%
mutate(
group_level = ifelse(group == "Healthy", 0, 1)
)
current_protein_values <- protein_values %>%
select(
id = sample_id,
abundance = !!sym(selected_protein_log_std)
)
association_data <- samples_tested %>%
left_join(
current_protein_values,
by = "id"
) %>%
filter(
!is.na(abundance)
)
lm_results <- lm(
formula = "abundance ~ group_level + age + bmi + sex",
data = association_data
)
summary(lm_results)
##
## Call:
## lm(formula = "abundance ~ group_level + age + bmi + sex", data = association_data)
##
## Residuals:
## Min 1Q Median 3Q Max
## -1.3736 -0.3283 -0.1226 0.2698 6.4926
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -0.153188 0.320226 -0.478 0.6327
## group_level 0.093163 0.097110 0.959 0.3381
## age 0.009273 0.004196 2.210 0.0278 *
## bmi -0.010061 0.010184 -0.988 0.3240
## sex -0.039089 0.084630 -0.462 0.6445
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.7013 on 309 degrees of freedom
## Multiple R-squared: 0.02357, Adjusted R-squared: 0.01093
## F-statistic: 1.865 on 4 and 309 DF, p-value: 0.1165
:pencil2: Plot the association with all classes.
classes <- c("Class_1", "Class_2a", "Class_2b", "Class_L")
plot_data <- NULL
for (class in classes) {
samples_tested <- samples_details %>%
filter(
group == "Healthy" | group == class
) %>%
mutate(
group_level = ifelse(group == "Healthy", 0, 1)
)
current_protein_values <- protein_values %>%
select(
id = sample_id,
abundance = !!sym(selected_protein_log_std)
)
association_data <- samples_tested %>%
left_join(
current_protein_values,
by = "id"
) %>%
filter(
!is.na(abundance)
)
association_data$class <- class
plot_data <- rbind(plot_data, association_data)
}
ggplot() +
geom_hline(
yintercept = 0
) +
geom_point(
data = plot_data,
mapping = aes(
x = group_level,
y = abundance,
col = class
),
alpha = 0.2
) +
geom_smooth(
data = plot_data,
mapping = aes(
x = group_level,
y = abundance,
col = class
),
formula = "y ~ x",
method = "lm"
) +
scale_x_continuous(
name = "Case or Control"
) +
scale_y_continuous(
name = "Protein abundance"
) +
facet_grid(
. ~ class
) +
theme(
legend.position = "none"
)
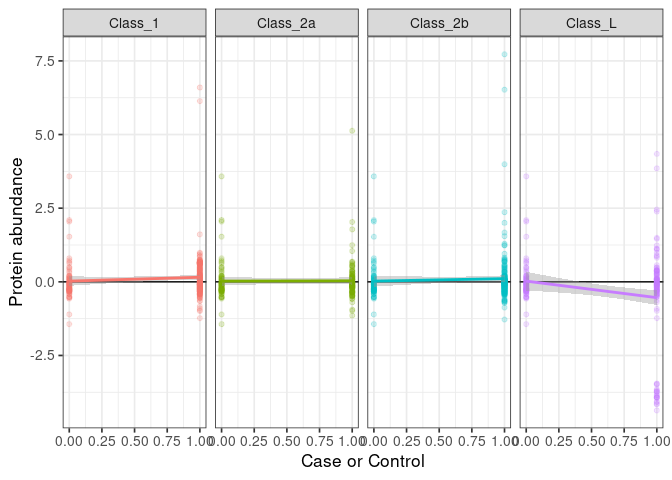
:pencil2: For every protein, run a linear model of every patient class vs healthy taking age, BMI, and sex as covariate.
classes <- c("Class_1", "Class_2a", "Class_2b", "Class_L")
n <- length(classes) * nrow(protein_details)
results <- data.frame(
class = character(n),
protein_id = character(n),
uniprot_id = character(n),
protein_name = character(n),
beta = numeric(n),
se = numeric(n),
p = numeric(n),
n = numeric(n),
stringsAsFactors = F
)
i <- 1
for (class in classes) {
print(glue("{Sys.time()} - Processing {class}"))
samples_tested <- samples_details %>%
filter(
group == "Healthy" | group == class
) %>%
mutate(
group_level = ifelse(group == "Healthy", 0, 1)
)
for (protein_index in 1:nrow(protein_details)) {
protein_id <- protein_details$protein_id[protein_index]
uniprot_id <- protein_details$uni_prot[protein_index]
protein_name <- protein_details$target_full_name[protein_index]
col_name <- paste0(protein_id, "_log_std")
current_protein_values <- protein_values %>%
select(
id = sample_id,
abundance = !!sym(col_name)
)
association_data <- samples_tested %>%
left_join(
current_protein_values,
by = "id"
) %>%
filter(
!is.na(abundance)
)
lm_results <- lm(
formula = "abundance ~ group_level + age + bmi + sex",
data = association_data
)
lm_summary <- summary(lm_results)
results$class[i] <- class
results$protein_id[i] <- protein_id
results$uniprot_id[i] <- uniprot_id
results$protein_name[i] <- protein_name
results$beta[i] <- lm_summary$coefficients["group_level", 1]
results$se[i] <- lm_summary$coefficients["group_level", 2]
results$p[i] <- lm_summary$coefficients["group_level", 4]
results$n[i] <- nrow(association_data)
i <- i + 1
}
}
## 2022-06-20 17:42:34 - Processing Class_1
## 2022-06-20 17:45:38 - Processing Class_2a
## 2022-06-20 17:48:39 - Processing Class_2b
## 2022-06-20 17:51:34 - Processing Class_L
:pencil2: Plot the significance against the effect size for each comparison.
ggplot() +
geom_point(
data = results,
mapping = aes(
x = beta,
y = -log10(p),
col = class
),
alpha = 0.2
) +
geom_hline(
yintercept = 2,
col = "darkgreen",
linetype = "dashed"
) +
scale_x_continuous(
name = "Effect size"
) +
scale_y_continuous(
name = "p-value [-log10]"
) +
facet_grid(
. ~ class
) +
theme(
legend.position = "none"
)
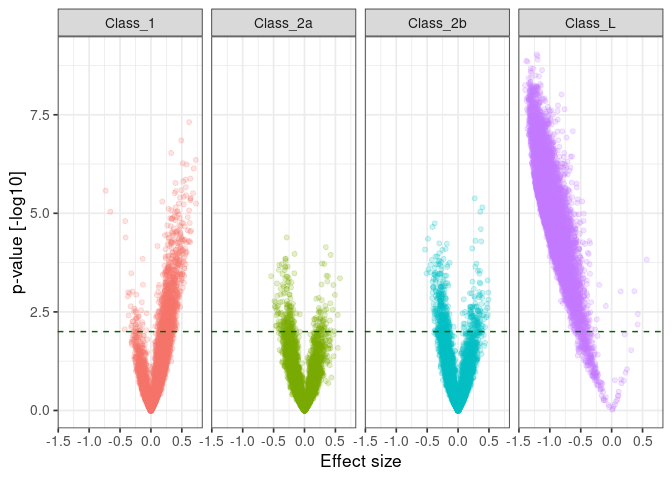
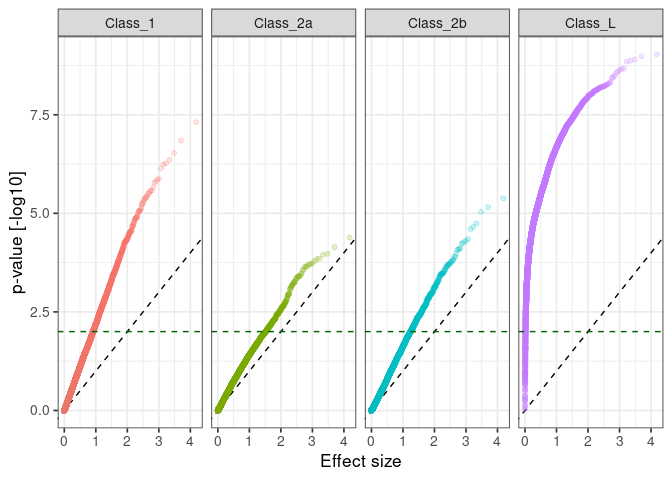
:pencil2: Plot the observed against expected p-value for each class.
qq_data <- results %>%
group_by(
class
) %>%
mutate(
logP = -log10(p)
) %>%
arrange(
logP
) %>%
mutate(
expectedLogP = sort(-log10(ppoints(n = n())))
) %>%
ungroup()
ggplot() +
geom_abline(
slope = 1,
intercept = 0,
color = "black",
linetype = "dashed"
) +
geom_point(
data = qq_data,
mapping = aes(
x = expectedLogP,
y = logP,
col = class
),
alpha = 0.2
) +
geom_hline(
yintercept = 2,
col = "darkgreen",
linetype = "dashed"
) +
scale_x_continuous(
name = "Effect size"
) +
scale_y_continuous(
name = "p-value [-log10]"
) +
facet_grid(
. ~ class
) +
theme(
legend.position = "none"
)