- Introduction
- 1. Background
- 2. Retrieving sequences
- 3. Sequence alignment
- 4. Distance-based analyses
- 5. Recombination
- 6. Maximum likelihood based analyses
- 7. Visualising trees
- 8. Time-stamped phylogenies
- 9. Bayesian reconstruction of time trees
- 10. Effective population size estimation
- 11. Structured populations
- 12. References
- Published with GitBook
Theory
Introduction
- Methods based on distances are typically very fast compared to model-based methods that will be discussed later
- Generating a distance matrix
- Constructing a tree by neighbour joining
- Generating a network of 'clustered' sequences
Generating a distance matrix
- Given a multiple sequence alignment, there are several models of DNA evolution that have been proposed e.g.
- JC69
- HKY85
- TN93
- GTR
- See Wikipedia for a nice description of these
Distance calculations
- An estimate of the distance between pairs of sequences can be obtained using a closed expression for several of these models
The Tamura-Nei (1993) model
- Unequal base frequencies
- Different rates of A-G and C-T transitions- The transversion rate can be different from the transition rate
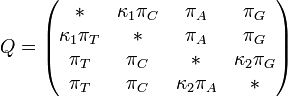
Source: Wikipedia
Neighbour joining
- Given a distance matrix, the neighbour joining algorithm builds a tree
- Identify the closest neighbours using information from the distance matrix
- Join neighbours
- Calculate distance of taxa in the pair to new node
- Calculate distance of taxa outside the pair to the node
- Replace joined neighbours with new node and repeat
NJ algorithm

Measuring error
- Neighbour joining produces a single tree, with no assessment of goodness-of-fit, or any idea of confidence in the tree
- A common approach to obtain a confidence set of trees is to bootstrap the sequences
Bootstrapping
- Classical statistical inference is based on the concept of hypothetical repetitions
- Bootstrapping involves random resampling of the data with replacement
- For sequence data, by generating new datasets from the old one, by resampling the columns of the sequence alignment
Clustering
- While a tree may capture 'deep' evolutionary relationships