- Introduction
- 1. Background
- 2. Retrieving sequences
- 3. Sequence alignment
- 4. Distance-based analyses
- 5. Recombination
- 6. Maximum likelihood based analyses
- 7. Visualising trees
- 8. Time-stamped phylogenies
- 9. Bayesian reconstruction of time trees
- 10. Effective population size estimation
- 11. Structured populations
- 12. References
- Published with GitBook
Practice
Prerequisites
Load libraries
library(ape)
library(stepwise)
library(magrittr)
library(kdetrees)
library(phangorn)
source("recombination.R")
In addition to the libraries, I have also written some functions:
slidingWindowAlignment: generates sliding window alignmentssbtest: generates trees either side of a breakpoint, and calculates the distance between the treesdisttree: calculates distances between all trees in a list
Set filename and outgroup
seqfilename <- "CRF7.fas"
outgroup <- "J_SE7887"
Read FASTA file
seqdata <- read.dna(seqfilename,format="fasta",as.matrix=TRUE)
seqdata
## 10 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 7894
##
## Labels: C_C2220 CRF7_C54A A_92UG037 B_JRFL D_NDK F_93BR020 ...
##
## Base composition:
## a c g t
## 0.366 0.173 0.236 0.225
Output data with 'clean' sequence names in Phylip format
Some of the tests we will use need to have sequence data in Phylip format, so we use R to convert.
seqdata.stepwise <- seqdata
row.names(seqdata.stepwise) <- makeLabel(row.names(seqdata),len=9)
write.dna(seqdata.stepwise,file=paste(seqfilename,".stepwise",sep=""),format="interleaved",colsep="",nbcol=-1)
Test for recombination using Maxchi
seqdata.maxchi <- maxchi(paste(seqfilename,".stepwise",sep=""),breaks=seg.sites(seqdata),winHalfWidth=50,permReps=100)
summary(seqdata.maxchi)
## --------------------------------------------------
## There were 12 site-specific MaxChi statistics significant at the
## 10 percent level (90th percentile = 21.007, 95th percentile = 22.243):
##
## Number Location MaxChi pairs
## 1 293 23.645* (B_JRFL :G_92NG083 )
## 2 294 21.583* (B_JRFL :G_92NG083 )
## 3 297 23.377* (B_JRFL :G_92NG083 )
## 4 300 25.253* (B_JRFL :G_92NG083 )
## 5 301 23.377* (B_JRFL :G_92NG083 )
## 6 387 21.236* (B_JRFL :G_92NG083 )
## 7 6022 23.188* (CRF7_C54A :G_92NG083 )
## 8 6084 21.374* (C_C2220 :G_92NG083 )
## 9 6085 23.377* (C_C2220 :G_92NG083 )
## 10 6090 21.374* (CRF7_C54A :G_92NG083 )
## 11 6091 23.188* (CRF7_C54A :G_92NG083 )
## 12 6092 21.374* (C_C2220 :G_92NG083 )
## --------------------------------------------------
## Notes - "Location" is the polymorphic site just before the proposed breakpoint.
## - MaxChi statistics significant at the 5 percent level indicated by a *
Test for recombination using phylogenetic profiling
Running the same window and permutations using phylogenetic profiling.
seqdata.phylpro <- phylpro(paste(seqfilename,".stepwise",sep=""),breaks=seg.sites(seqdata),winHalfWidth=50,permReps=100)
summary(seqdata.phylpro)
## Length Class Mode
## 0 NULL NULL
Test for recombination using a sliding window phylogeny
We have already covered making a tree with a sequence alignment using a distance based approach, which involves making a distance matrix, performing tree reconstruction, perhaps rooting the tree, and plotting it out.
Making a set of trees
As the sequences are stored in a matrix, it is straightforward to make a list of alignments, each of which is a window on the original alignment.
The following command generates sequence alignments 300 base pairs long, moving in steps of 10.
seqdata.slide <- slidingWindowAlignment(seqdata,300,10)
length(seqdata.slide)
## [1] 760
This generates a list of alignments. In R, there is a command, lapply, that applies a command to each element in a list. The following generates a list of distance matrices, then a list of neighbour joining trees.
seqdata.slide.nj <- lapply(seqdata.slide,dist.dna,model="TN93",as.matrix=TRUE) %>%
lapply(.,njs)
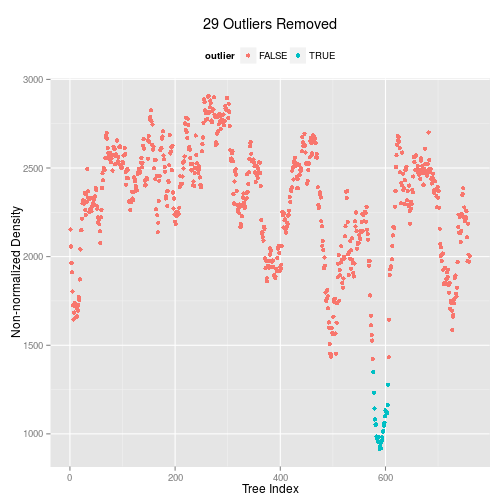
Outlier detection
We now have a list of trees. How do we determine whether a single tree explains all the sub-alignments? One approach is to work out whether there are 'outlying trees'. The command kdetrees computes a distribution of trees, then determines whether there are trees in the 'tail' of the distribution. This requires an outgroup sequence to root the tree.
seqdata.slide.nj.kde <- kdetrees(seqdata.slide.nj,outgroup=outgroup)
seqdata.slide.nj.kde
## Call: kdetrees(trees = seqdata.slide.nj, outgroup = outgroup)
## Density estimates:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 915.4 2071.0 2364.0 2272.0 2531.0 2906.0
## Cutoff: 1380.953
##
## Outliers detected:
## [1] 577 578 579 580 581 582 583 584 585 586 587 588 589 590 591 592 593
## [18] 594 595 596 597 598 599 600 601 602 603 604 605
Plotting out the output from this function will show the outlying trees, as a function of the index of the tree - each of which represents the tree from a slice of the original alignment.
plot(seqdata.slide.nj.kde)
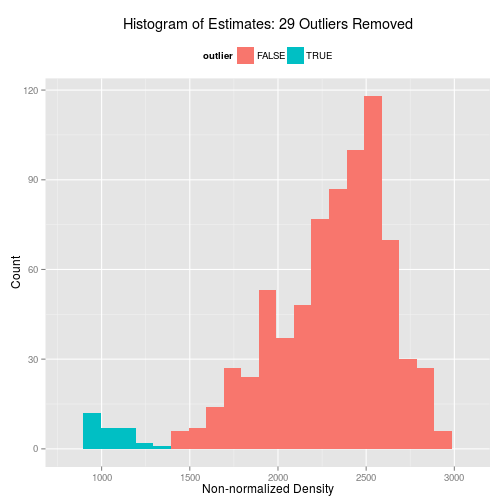
We can also plot out a histogram.
hist(seqdata.slide.nj.kde)
Lets take two trees, one outlier, and one non-outlier.
par(mfrow=c(1,2))
plot(seqdata.slide.nj[[200]])
plot(seqdata.slide.nj[[600]])
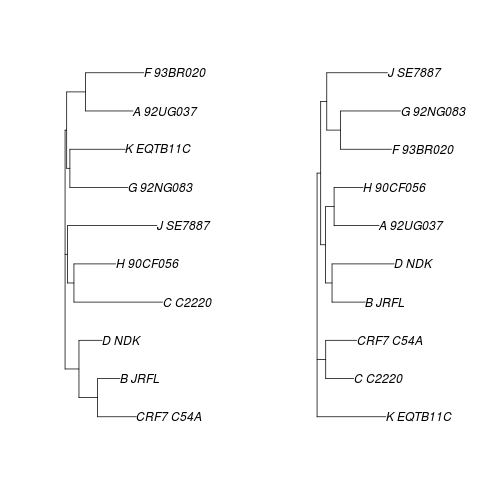
How are these different?
The command cophyloplot in ape allows us to plot two trees face-to-face in order to compare them more easily. This function takes the two trees as arguments, plus a matrix with the associations between the labels in the two trees - this is for cases where the labels in the trees may be different (e.g. comparing host and parasite phylogenies). In our case, the labels are the same.
association <- matrix(c("CRF7_C54A","CRF7_C54A"),nrow=1,ncol=2)
cophyloplot(seqdata.slide.nj[[200]],seqdata.slide.nj[[600]],assoc=association)
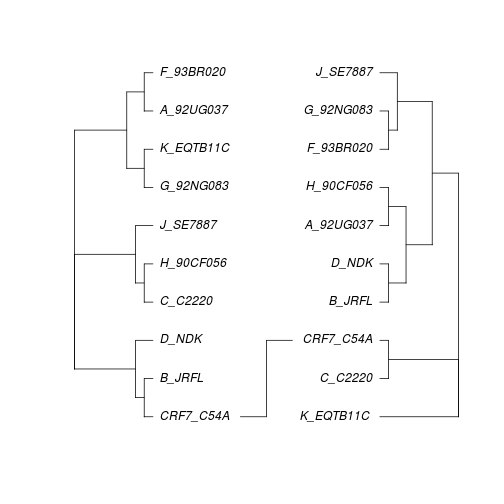
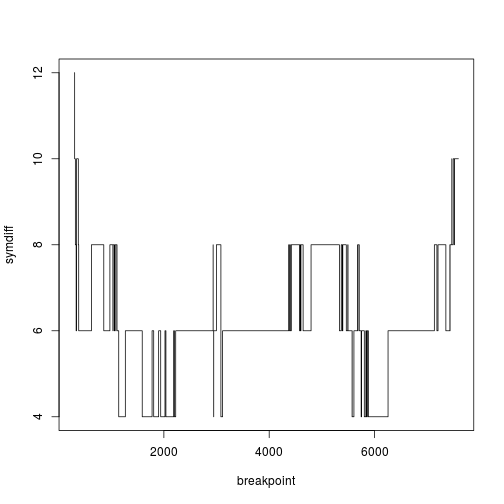
Single breakpoint test
A sliding window approach may be best if there are recombinant fragments, but if the sequences have a single recombinant breakpoint, then a different approach may be more powerful. This function splits the alignment into two (at least 300 base pairs long), and calculates the distance between the trees for either side of the breakpoint.
seqdata.sbt <- sbtest(seqdata,300,"TN93")
Now we can plot the distance between the trees as a function of the breakpoint.
plot(symdiff~breakpoint,data=seqdata.sbt,type="s")